

Publication
Research Article
International Journal of MS Care
Effect of Alemtuzumab Infusions on Vital Signs
Abstract
Background:
Alemtuzumab efficacy and safety were established in phase 3 randomized trials. We characterize vital signs during and after the first alemtuzumab infusion course.
Methods:
Patients with relapsing-remitting multiple sclerosis commercially prescribed alemtuzumab 12 mg/day on 5 consecutive days (initial course) were enrolled in this prospective, observational study. Preinfusion medications included methylprednisolone, antihistamine, and antipyretics. Primary end point: change from precourse baseline in vital signs during and 2 hours after each alemtuzumab infusion. Secondary end points: infusion duration and serious adverse events (AEs) starting within 24 hours and within 7 days after infusion (AEs collected up to 15 days after treatment). Potentially clinically significant vital sign abnormalities were based on predefined thresholds from literature review.
Results:
In the 304 patients treated, minimal increases in mean systolic (≤8 mm Hg) and diastolic (≤3 mm Hg) blood pressures from precourse baseline were observed on infusion days 3 to 5. An increase in mean heart rate (20 beats per minute) during the first infusion day normalized by day 2, and smaller increases (5 beats per minute) occurred during subsequent infusions. Serious AEs occurred in two patients (0.7%) during or within 24 hours after infusion and in three patients (1.0%) within 7 days. Mean/median infusion duration was 4 hours. Vital sign abnormalities with potential clinical significance occurred in 62.5% of patients.
Conclusions:
Although most patients had potentially clinically significant vital sign abnormalities, mean changes from baseline during and after infusion of the first alemtuzumab course were clinically insignificant. No new safety signals were detected.
Alemtuzumab (Lemtrada; Sanofi Genzyme) is a humanized, anti-CD52 monoclonal antibody indicated for the treatment of patients with relapsing forms of multiple sclerosis (MS), and it is licensed in more than 65 countries.1 The approved dosing schedule comprises two courses: intravenous (IV) infusion of 12 mg/day on 5 consecutive days (course 1) and on 3 consecutive days 12 months later (course 2). Up to two additional courses may be given to patients with disease activity.1 In the pivotal phase 3 Comparison of Alemtuzumab and Rebif Efficacy in Multiple Sclerosis (CARE-MS) I and II trials in patients with relapsing-remitting MS (RRMS), alemtuzumab significantly reduced relapses and brain volume loss over 2 years and reduced the risk of 6-month disability worsening compared with subcutaneous interferon beta-1a (Rebif; EMD Serono).2 3 An increased rate of confirmed disability improvement was also seen in patients from CARE-MS II.3 Alemtuzumab efficacy was maintained over 6 years in an extension study, during which 63% (CARE-MS I) and 50% (CARE-MS II) of patients received no additional alemtuzumab courses or other disease-modifying therapies.4–6 Longer-term outcomes are being collected in a second extension study (TOPAZ).
The most frequent adverse events (AEs) in the phase 3 trials were infusion-associated reactions (IARs), defined as any AE that occurred during infusion or within the following 24 hours. The IARs were mostly mild or moderate and most commonly manifested as headache, rash, or pyrexia. The incidence of IARs declined with each treatment course. Data from 5 years of follow-up show that during course 1, the IAR incidence was 84.7%, decreasing to 68.6%, 64.6%, and 62.8% for courses 2, 3, and 4, respectively.7 Serious IAR incidence was 2% or less with each course. Serious AEs (SAEs) related to vital signs each occurred in less than 1% of patients (pyrexia, atrial fibrillation, and hypotension); nonserious tachycardia occurred in less than 5% of patients. The IARs were generally managed with monitoring for early detection of events, prompt symptomatic treatment, and, if necessary, slowing or stopping the infusion.7 8
During the clinical development program, vital signs were not recorded systematically during and after alemtuzumab infusions. The Alemtuzumab Infusion Vital Signs Study was designed to further characterize the safety profile of alemtuzumab by monitoring vital signs during and after infusion of course 1.
Methods
Patients
The Alemtuzumab Infusion Vital Signs Study was a prospective, observational cohort study conducted at 32 sites in the United States from November 1, 2015, to March 31, 2017. Key eligibility criteria included diagnosis of RRMS, planned treatment with alemtuzumab as per the approved local label and independent of entry in the study, and signed informed consent. The key exclusion criteria included previous alemtuzumab treatment and any illness or condition that would compromise the patient’s safety or the patient’s ability to understand patient information, provide informed consent, comply with the study protocol, or complete the study or that would interfere with interpretation of the results.
Procedures and End Points
Patients were screened for eligibility within 30 days of the first alemtuzumab infusion and were followed up by visit or telephone 7 to 15 days after the last infusion day of course 1. Total participation time for each patient, including screening, treatment with alemtuzumab course 1, and follow-up, was 2 to 7 weeks. Alemtuzumab 12 mg/day was administered by IV infusion over 4 hours on 5 consecutive days. The duration of infusion was extended if required at the discretion of the investigator. Premedication for all patients included 1 g of IV methylprednisolone on the first 3 infusion days, as well as antihistamines and antipyretics on each infusion day. The antihistamines and antipyretics routinely administered in these patients included diphenhydramine (25–200 mg orally, 25–50 mg IV, or 25–50 mg intramuscularly), famotidine (10–40 mg orally or 20 mg IV), ranitidine (50–300 mg orally or 50 mg IV), cetirizine (10 mg orally), hydroxyzine (25–150 mg orally), paracetamol (500–3000 mg orally), ibuprofen (200–1000 mg orally), naproxen (220–500 mg orally), and acetylsalicylic acid (81–325 mg orally). Additional antihistamine, antipyretic, or other symptomatic medications could be given during infusion at the investigator’s discretion. Patients also received acyclovir (200 mg orally twice daily) for herpetic viral infection prophylaxis beginning on the first day of course 1 and continuing for 2 months after completion or until the CD4+ count was at least 200 mL/μL. On infusion days, infusion duration, premedication, prophylaxis, and symptomatic treatments were recorded. Vital signs such as systolic and diastolic blood pressure (BP), heart rate, respiratory rate, and temperature were monitored before methylprednisolone treatment, immediately before each alemtuzumab infusion, and hourly from the start of alemtuzumab infusion until 2 hours or more after infusion. Any AEs were recorded from the first infusion until the follow-up visit 7 to 15 days after the last infusion of alemtuzumab course 1.
The primary end point was change in vital signs during alemtuzumab infusion and the 2-hour postinfusion observation period with respect to precourse baseline. Precourse baseline was defined as the value of the vital sign immediately before alemtuzumab infusion on the first infusion day, whereas preinfusion baseline was defined as the value of the vital sign immediately before alemtuzumab infusion on each infusion day. Secondary end points included the incidence of SAEs during or within 24 hours after infusion, the incidence of SAEs during or within 7 days after infusion, and the duration of infusion. Potentially clinically significant abnormalities were defined as heart rate of 50 beats per minute (bpm) or less or 120 bpm or greater (under conditions of no exercise); systolic BP (SBP) of 95 mm Hg or less and a 20–mm Hg or greater decrease from baseline (low) or 160 mm Hg or greater (high); diastolic BP (DBP) of 40 mm Hg or less and a 10–mm Hg or greater decrease from baseline (low) or 110 mm Hg or greater and a 10–mm Hg or greater increase from baseline (high). Adverse events of special interest included autoimmune AEs (nephropathies, cytopenias, thyroid disorders, and immune thrombocytopenia), serious infections, pneumonitis, pregnancy (including healthy pregnancy), and malignancies.
The local institutional review board/independent ethics committee approved the protocol, and all the patients provided written informed consent.
Statistical Analysis
A sample size of 300 patients was planned to enable detection of events at an incidence of 0.33% with an exact 95% CI of 0.01% to 1.84% and an upper bound of 1.22% for incidence of events that did not occur in any patient under study. Descriptive statistical analysis was performed on all vital signs at each time point and for change in vital signs from precourse baseline at each time point. For patients with missing baseline vital sign values, the values at the time before methylprednisolone treatment were used. No imputation was made for patients with missing vital sign values at any postbaseline time points.
Results
Patient Characteristics and Infusion Duration
Of 307 patients screened, 305 (99.3%) met the inclusion criteria and were enrolled in the study, and 304 of them (99.7%) were treated (Figure S1, which is published in the online version of this article at ijmsc.org). At baseline, patients had a mean disease duration of 10.5 years; within the past 2 years, 96.7% had received therapy and 56.3% had experienced relapse activity (Table S1). No patients discontinued study participation, and 269 (88.5%) completed treatment within 5 to 7 calendar days of the first alemtuzumab infusion. The mean and median infusion durations were both 240 minutes (4 hours; range, 50–437 minutes); 1.6% to 3.9% of patients on any given treatment day exceeded 4 hours of infusion.
Vital Signs
During course 1 of alemtuzumab infusion, there were minimal mean BP changes during and after infusion on any treatment day with respect to precourse baseline (Figure 1A and B). There were mean decreases of less than 5 mm Hg from precourse baseline in SBP on day 1 and in DBP on days 1 and 2. Mean increases in SBP of 8 mm Hg or less and in DBP of 3 mm Hg or less from precourse baseline were noted at preinfusion baseline on days 3 to 5, which persisted through the last observation of each day.


Changes from precourse baseline in (A) systolic blood pressure, (B) diastolic blood pressure, (C) heart rate, (D) respiratory rate, and (E) body temperature during alemtuzumab infusion
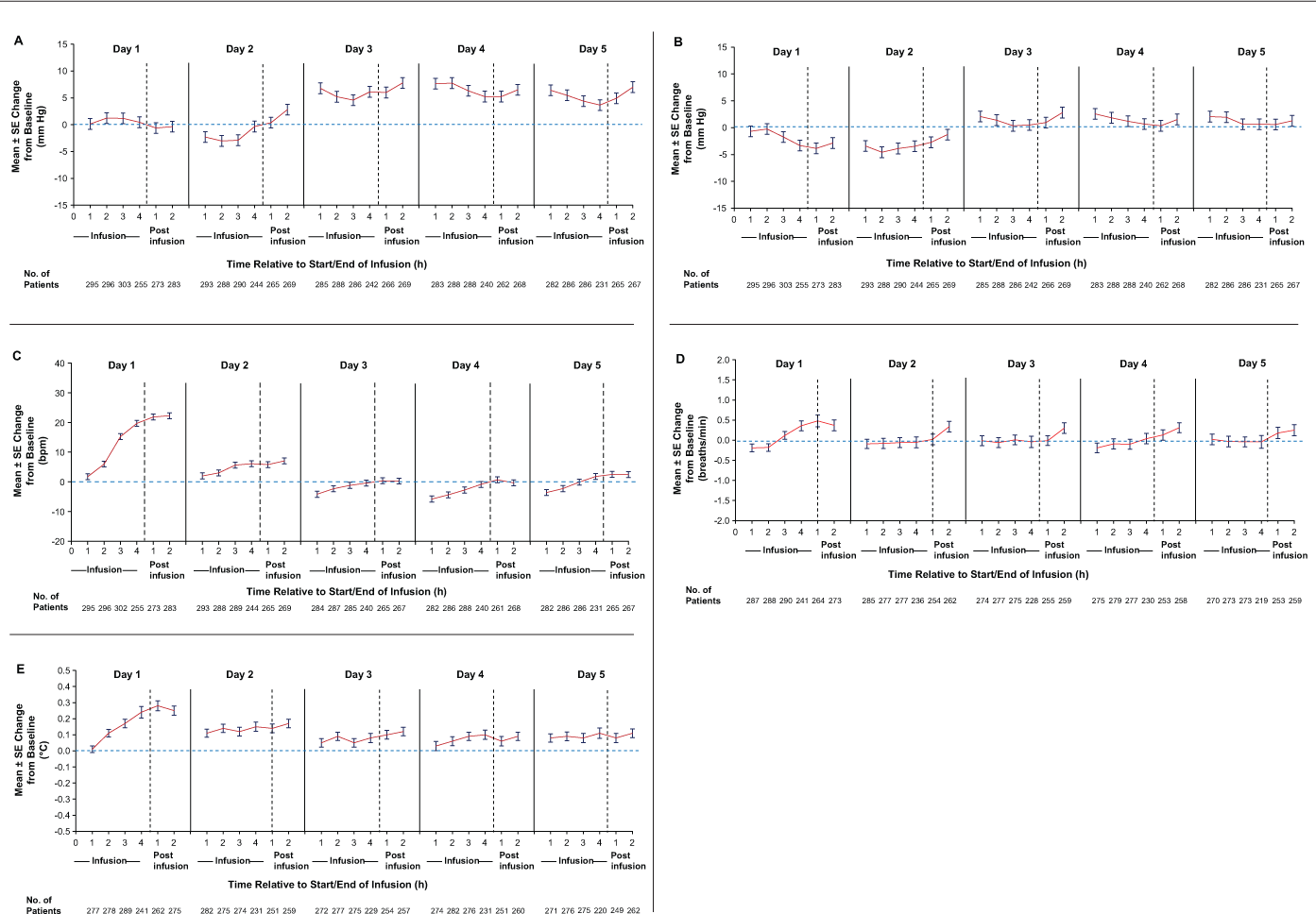
On the first day of alemtuzumab infusion (day 1), a gradual increase in mean heart rate of approximately 20 bpm with respect to precourse baseline was observed; this increase persisted after the infusion (Figure 1C). On days 2 to 5, mean increases of approximately 5 bpm were detected during infusion. On days 3 to 5, the preinfusion baseline heart rate was 5 bpm or less lower than the precourse baseline value. The mean heart rate remained within normal limits throughout and after each infusion (60–100 bpm).
A gradual mean increase in respiratory rate of approximately 0.5 breaths per minute from precourse baseline was observed during infusion on day 1 (Figure 1D). The increase persisted after infusion but normalized by day 2. The respiratory rate remained stable from day 2 to day 5 during the 4-hour infusion period, with a slight transient increase (range of mean change, −0.01 to 0.34 breaths per minute) during the postinfusion period. Overall, the mean change in respiratory rate from precourse baseline was not clinically meaningful.
During the 4-hour infusion period on day 1 there was a gradual mean increase in basal body temperature of less than 0.5°C (<0.9°F) from precourse baseline. This increase persisted after infusion, normalized by day 2, and remained at baseline level throughout subsequent infusions (Figure 1E).
Few patients (<4% on any given day) received infusions of more than 4 hours; vital signs at hours 5, 6, and 8 are presented separately because these data are highly variable due to small sample size and bias toward patients with prolonged infusion due to IARs (Table S2). Mean change from precourse baseline to hours 5, 6, and 8 in each vital sign varied from the mean change noted at earlier time points.
Adverse Events
The incidence of overall alemtuzumab treatment-emergent AEs was 71.7% (Table 1). All reported AEs occurred within 15 days of the last alemtuzumab infusion. The AEs were mostly mild (62.8% of patients) to moderate (30.9%) in severity; 3% of patients had severe AEs. The AEs were most commonly reported as headache, nausea, fatigue, pain, rash, pyrexia, flushing, and insomnia. Nine treatment-emergent SAEs were reported in eight patients (2.6%), with two patients (0.7%) having SAEs within 24 hours after any alemtuzumab infusion (pyrexia, pneumonia). The first patient experienced grade 1 pyrexia with a body temperature of 101.6°F on the day of the fifth alemtuzumab infusion. He was treated with acetaminophen, ibuprofen, IV fluids, and hydration, and he recovered 3 days later. The second patient had bilateral pneumonia, was treated with meropenem, and was transitioned to levofloxacin after negative evaluation results. Further alemtuzumab infusion was postponed for 5 days; the patient recovered from pneumonia 7 weeks after onset. Three patients had SAEs within 7 days after any alemtuzumab infusion (muscle spasms, asthenia and peripheral edema, fecal impaction), and the remaining three (1.0%) SAEs occurred between 7 and 15 days after the last infusion (bronchopulmonary aspergillosis, colon cancer, sepsis). Of these, the bronchopulmonary aspergillosis, muscle spasms, and pyrexia were deemed to be related to alemtuzumab, whereas the sepsis, colon cancer, pneumonia, fecal impaction, and asthenia and peripheral edema were considered to be unrelated by the investigator. The patient diagnosed as having bronchopulmonary aspergillosis received ceftriaxone, meropenem trihydrate, and oral voriconazole as a corrective treatment and recovered from the condition 5 months after the last infusion of alemtuzumab. The patient with colon cancer underwent segmental resection of the colon with regional lymphadenectomy and recovered from the event 54 days after the last infusion of alemtuzumab. The sepsis case was fatal; the patient was a 77-year-old man with an immediate treatment history of fingolimod with no washout. He was reported to have grade 3 sepsis 10 days after the last infusion of alemtuzumab and tested positive for Citrobacter koseri. He was treated with piperacillin/tazobactam, vancomycin, and levofloxacin, but he died of septic shock 15 days after the last infusion of alemtuzumab. Four patients (1.3%) had AEs of special interest, including those with bronchopulmonary aspergillosis, colon cancer, and sepsis; a pregnancy with onset 11 days after the last alemtuzumab infusion was also reported as an AE of special interest and resulted in a healthy infant.


Adverse events in patients receiving alemtuzumab infusions
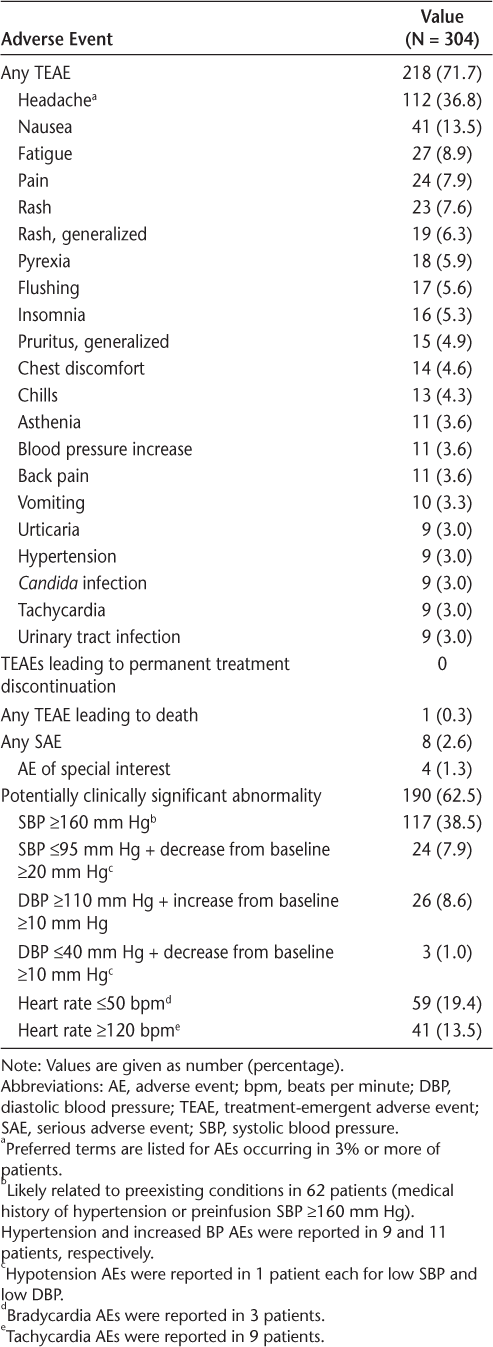
During alemtuzumab infusion, 1354 potentially clinically significant abnormalities were reported in 190 patients (62.5%). High SBP (≥160 mm Hg) was the most common (38.5%) (Table 1). Approximately half of the patients (n = 62) with high SBP had a medical history of hypertension or had preinfusion SBP of 160 mm Hg or higher. High SBP was reported as the AE of hypertension and increased BP in nine (3.0%) and 11 (3.6%) patients, respectively. High SBP was mainly observed on days 3 to 5 and was distributed intermittently before, during, and after infusion on each day. Mean peak SBP in patients with high SBP potentially clinically significant abnormalities was 176 mm Hg (range, 160–232 mm Hg). Other potentially clinically significant abnormalities included low SBP (≤95 mm Hg; 7.9%), high DBP (≥110 mm Hg; 8.6%), or low DBP (≤40 mm Hg; 1.0%). Hypotension AEs were reported in two patients (0.7%). Low heart rate (≤50 bpm; 19.4%) and high heart rate (≥120 bpm; 13.5%) potentially clinically significant abnormalities were infrequently reported as AEs of bradycardia (n = 3; 1.0%) and tachycardia (n = 9; 3.0%), respectively. None of the AEs related to BP or heart rate qualified as serious.
Discussion
This observational study showed that for most patients, alemtuzumab infusion was associated with minimal, transient, and clinically insignificant changes in vital signs. Overall, SAEs occurred in less than 3% of patients, and no new safety signals were observed.
The data in the present study are consistent with those of Thomas et al,9 who reported that in patients with RRMS (n = 15), infusion of alemtuzumab 12 mg did not have any significant effect on BP or body temperature. In that study, patients were given a similar premedication regimen as patients in the present study. The transient increases in heart rate observed herein, particularly on day 1, are also consistent with those reported by Thomas et al and may be related, at least in part, to methylprednisolone pretreatment given on the first 3 days of alemtuzumab infusions.9 10 Glucocorticoids are known to have cardiovascular effects, including modulation of vascular tone and cardiac output.11 Cases of cardiac arrhythmias, including sinus bradycardia, atrial fibrillation, and tachycardia, in response to glucocorticoids have also been reported.12 13 In the present study, the largest heart rate increase coincided with methylprednisolone treatment; however, heart rate increases moderated on the subsequent 2 days, on which methylprednisolone was also administered. The BP results did not show changes that were temporally related to methylprednisolone administration. Mean heart rate and BP values remained within normal limits throughout the infusion period despite transient fluctuations.
Fifty-three percent of patients with potentially clinically significant abnormalities related to BP had a medical history of hypertension or had high BP at baseline. This finding suggests that patients with preexisting high BP should be monitored carefully during alemtuzumab infusion to rapidly detect BP changes and enable prompt intervention if necessary.
Mean SBP remained elevated (7 mm Hg) 2 hours after the last infusion. Because vital signs were not recorded after the last infusion day, it is unclear how long this increase in BP persisted; however, in the phase 3 trials, there was no evidence of BP elevation at the 30-day postinfusion visit.
The slight changes in body temperature during infusion and in respiratory rate after infusion, particularly on day 1, normalized by the next infusion day. Temperature changes may have been minimized by antipyretic premedication; peak temperature on day 1 occurred 5 hours or more after premedication, temporally consistent with the duration of effect of paracetamol. Respiratory rate and temperature during and after infusion were within normal limits; changes in these parameters were not clinically meaningful.
The alemtuzumab IARs in the present study were similar to those in the previous alemtuzumab clinical trials.7 8 In the CARE-MS studies, patients who received alemtuzumab 12 mg (N = 811) most frequently had IARs of rash, headache, pyrexia, urticaria, nausea, pruritus, and flushing.7 However, for some IARs, incidences in the pivotal trials were higher than observed herein, including rash (36% vs 8%), pyrexia (15% vs 6%), and urticaria (12% vs 3%).7 The reason for these differences may be related to the antihistamine and antipyretic premedication required under this protocol; in the CARE-MS studies, these premedications were permitted but not required. Also note that the pivotal trials defined IARs as AEs occurring within 24 hours after infusion, whereas the present study reported AEs within 15 days after the last infusion.2 3
In addition to the short-term changes in vital signs, patients treated with alemtuzumab should be monitored long-term, particularly for autoimmune events. In phase 2 and 3 core and extension trials, respectively, patients most commonly developed thyroid events (42%) and, less frequently, immune thrombocytopenia (2.2%) and nephropathies (0.34%) over a median of 6.1 years of follow-up after the first infusion.14–16 A monitoring program has been used to detect and manage these AEs to minimize the associated risks.17
Limitations of the study revolve around its observational design. There were no control arms to separate the effects of alemtuzumab infusion from the effects of methylprednisolone pretreatment or to make direct observations about the effectiveness of antihistamine and antipyretic premedication in preventing AEs. Furthermore, because the observation period of the study was limited to alemtuzumab course 1, it is unknown whether vital signs would differ during course 2, 12 months after the previous alemtuzumab exposure.
In summary, changes in vital signs of patients with RRMS treated with course 1 of alemtuzumab were generally determined to be clinically insignificant, and except for SBP, changes observed on days 1 through 4 recovered to baseline or near baseline by the subsequent infusion day. The AEs were infrequently serious. These data further characterize and support the manageable safety profile of alemtuzumab.
PRACTICE POINTS
Vital sign changes in patients with relapsing-remitting MS treated with the first course of alemtuzumab were clinically insignificant, and no new safety signals were detected.
A transient increase in heart rate was observed on the first infusion day.
Use of antihistamine and antipyretic premedication may reduce infusion-associated reactions.
All patients should be monitored carefully during alemtuzumab infusion; patients with preexisting elevated blood pressure may be predisposed to having further increases in blood pressure during treatment.
Acknowledgments
The authors and Sanofi thank the patients for their participation in the study and acknowledge the Alemtuzumab Infusion Vital Signs Study Steering Committee. The manuscript was reviewed for scientific accuracy by Darren P. Baker, PhD, Ericka M. Bueno, PhD, and Colin Mitchell, PhD, of Sanofi. Editorial support was provided by Valerie P. Zediak, PhD, and Noopur Mandrekar, PhD, of Envision Scientific Solutions and was funded by Sanofi.
References
Genzyme Therapeutics Ltd. Lemtrada™ (alemtuzumab 12 mg concentrate for solution for infusion): EU summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003718/WC500150521.pdf. Accessed March 12, 2018.
Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380:1819–1828.
Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380:1829–1839.
Coles AJ, Cohen JA, Fox EJ, et al. Alemtuzumab CARE-MS II 5-year follow-up: efficacy and safety findings. Neurology. 2017;89:1117–1126.
Havrdova E, Arnold DL, Cohen JA, et al. Alemtuzumab CARE-MS I 5-year follow-up: durable efficacy in the absence of continuous MS therapy. Neurology. 2017;89:1107–1116.
Ziemssen T, Thomas K. Alemtuzumab in the long-term treatment of relapsing-remitting multiple sclerosis: an update on the clinical trial evidence and data from the real world. Ther Adv Neurol Disord. 2017;10:343–359.
Mayer L, Fox E, LaGanke C, et al. Incidence of Infusion-Associated Reactions Decreases with Subsequent Courses of Alemtuzumab: 5-Year Data from the CARE-MS Extension Study. Paper presented at: 30th Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC); National Harbor, MD; June 1–4, 2016.
Caon C, Namey M, Meyer C, et al. Prevention and management of infusion-associated reactions in the Comparison of Alemtuzumab and Rebif (®) Efficacy in Multiple Sclerosis (CARE-MS) program. Int J MS Care. 2015;17:191–198.
Thomas K, Eisele J, Rodriguez-Leal FA, Hainke U, Ziemssen T. Acute effects of alemtuzumab infusion in patients with active relapsing-remitting MS. Neurol Neuroimmunol Neuroinflamm. 2016;3:e228.
Krieger S, Sorrells SF, Nickerson M, Pace TW. Mechanistic insights into corticosteroids in multiple sclerosis: war horse or chameleon? Clin Neurol Neurosurg. 2014;119:6–16.
McKay LI, Cidlowski JA. Physiologic and pharmalogical effects of corticosteroids. In: Kufe DW, Pollock RE, Weichselbaum RR, et al, eds. Holland-Frei Cancer Medicine. 6th ed. BC Decker; 2003.
Stroeder J, Evans C, Mansell H. Corticosteroid-induced bradycardia: case report and review of the literature. Can Pharm J. 2015;148:235–240.
Vasheghani-Farahani A, Sahraian MA, Darabi L, Aghsaie A, Minagar A. Incidence of various cardiac arrhythmias and conduction disturbances due to high dose intravenous methylprednisolone in patients with multiple sclerosis. J Neurol Sci. 2011;309:75–78.
Thomas K, Ziemssen T. Response to: S. Sega-Jazbec et al.: “Management of infusion related reactions associated with alemtuzumab in patients with multiple sclerosis” Multiple Sclerosis and Related Disorders 2017. Mult Scler Relat Disord. 2017;17:177–178.
Cuker A, Bass AD, Nadj C, et al; on behalf of the CAMMS223, CARE-MS I, CARE-MS II, and CAMMS03409 Investigators. Immune thrombocytopenia in alemtuzumab-treated MS patients: incidence, detection and management. Mult Scler. 2020;26:48–56.
Phelps R, Winston JA, Wynn D, et al. Incidence, management, and outcomes of autoimmune nephropathies following alemtuzumab treatment in patients with multiple sclerosis. Mult Scler. 2019;25:1273–1288.
Devonshire V, Phillips R, Wass H, Da Roza G, Senior P. Monitoring and management of autoimmunity in multiple sclerosis patients treated with alemtuzumab: practical recommendations. J Neurol. 2018;265:2494–2505.
Supplementary Material








